Project configuration¶
Sample dataset¶
To get you started, we provide two Diffusion Spectrum Imaging sample datasets. They already contain the correct folder structure described below. You can find the two raw datasets online:
project01_dsi
*connectome_0001* with timepoint *tp1* and DSI, T1 and T2 raw data
project02_dsi
*connectome_0002* with timepoint *tp1* and DSI, T1 raw data
If you produce any connectome dataset that you want to share with the community, we provide a curated cffdata repository on GitHub.
Run the Connectome Mapper¶
Now, you are ready to start the Connectome Mapper from the Bash Shell:
connectomemapper
Project configuration (folder structure)¶
Running the Connectome Mapper opens the main window as well as a menu toolbar on the top of the screen. The only enabled buttons are in the toolbar: the “New Connectome Data...” and “Load Connectome Data...” in the File menu.
If you have already configured a processing pipeline before, you can load the configuration by selecting the base directory using the “Load Connectome Data...” button. Otherwise, click “New Connectome Data...” and select the base directory for the project (i.e. the project that will contain all the processing steps and results for one subject). If you have several subject, we recommend to divide your project data into subject folders and timepoints. Selecting a folder will create the following folder structure:
├── myproject │ ├── subject001 │ │ └── tp1 <------ Your selected folder (base directory) │ │ ├── LOG │ │ ├── NIFTI │ │ ├── NIPYPE │ │ ├── RAWDATA │ │ │ ├── DSI │ │ │ ├── DTI │ │ │ ├── HARDI │ │ │ ├── T1 │ │ │ └── T2 │ │ ├── RESULTS │ │ └── STATS
You can also create the folder structure manually before selecting the base directory (existing folders won’t be overwritten).
Copy the diffusion (DSI, DTI, QBALL/HARDI) and morphological (T1, T2) images (DICOM series or single .nii.gz files) in the corresponding RAWDATA folders. The T2 images are optional but they improve the registration of the data. Do not put any other data is those folders besides T1, T2 or diffusion images.
Now you can click on “Check input data” button in the main window.
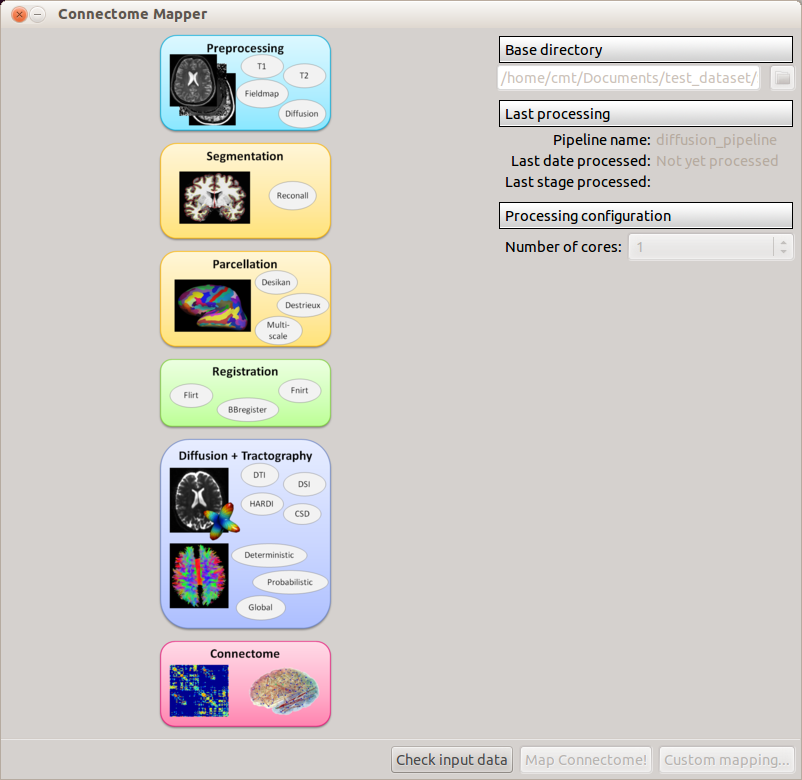
DICOM sequences will be converted to nifti format and nifti files copied into the NIFTI folder. A dialog box will appear to confirm the successful conversion. If several diffusion modalities are available, you’ll be asked to choose which modality to process.
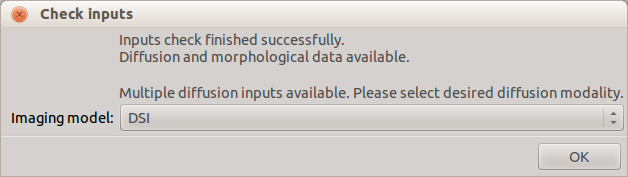
Once the diffusion modality is set, configuration of the pipeline is enabled. You can configure the processing stages by clicking on the respective buttons on the left. Pipeline information as base directory and last processing information are displayed on the right. You can also set the number of cores for multithreading the pipeline processing.
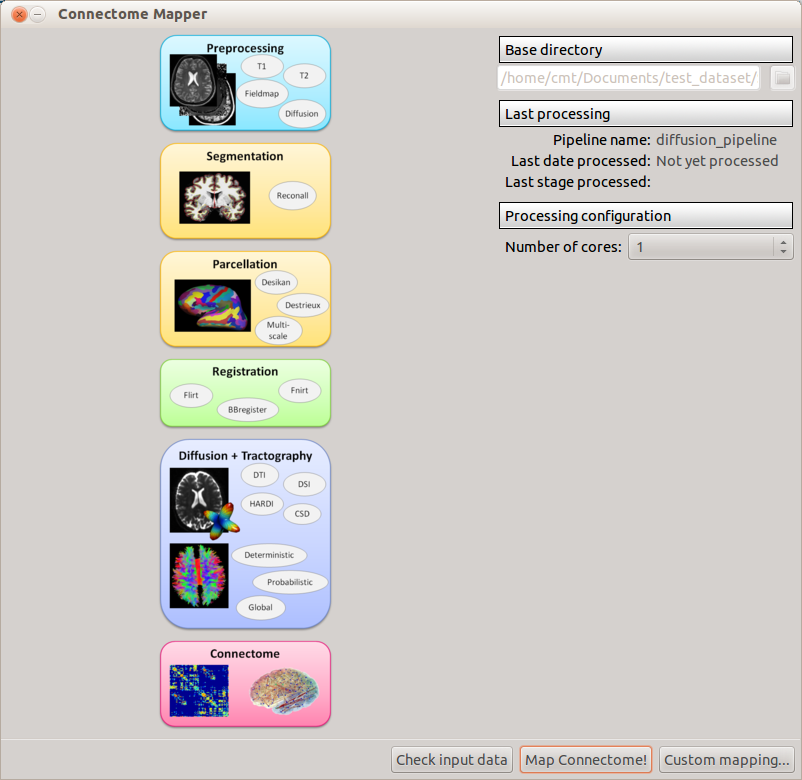
When the pipeline is configured, you can run the Map connectome! button. If you don’t want to process the whole pipeline at once, you can select which stage to stop at using the “Custom mapping...” button.
When the processing is finished, connectome tables will be saved in the RESULTS folder, in a subfolder named after the date and time the data was processed.
If you run into any problems or have any questions, post to the CMTK-users group.
Staring the pipeline without GUI¶
This can be useful if you want to automatically process different subjects or timepoints with the same configuration, or one subject with different configurations, etc...
Configure the pipeline as described previously, and instead of running it, save the configuration by clicking on the “Configuration” -> “Save configuration file...” button in the toolbar.
To run the analysis for a single subject, type:
connectomemapper input_folder config_file
To batch over a set of subject, you can make a bash script like this one:
#!/bin/bash
subjects_folders=(path/to/subject1/folder path/to/subject2/folder path/to/subject3/folder)
config_file = path/to/configfile.ini
for subject in "${subjects_folders[@]}"; do
connectomemapper "${subject}" "${config_file}"
done
Save the file as batch.sh and run it from the terminal:
./batch.sh